Vaccinegate: MRC-5 contenido en Priorix Tetra - Secuenciación completa del genoma

Estos últimos análisis fueron posibles gracias a la contribución activa de las asociaciones francesas. Asociación Liberté Informaciones Santé (ALIS) Ligue nationale pour la liberté des vacunaciones (LNPLV) y la Asociación Australiana Red Australiana de Riesgos de Vacunación (AVN)que agradecemos
La secuenciación de nueva generación se ha convertido en la herramienta preferida para el análisis en profundidad en el campo de la biología y las ciencias médicas, especialmente las de alta precisión. Gracias a estas herramientas, podemos abordar de una manera más moderna y completa una serie de aplicaciones como la secuenciación de novo, estudios metagenómicos y epigenómicos, secuenciación de transcriptomas y re-secuenciación de genomas.
Este último (re-secuenciación) se utiliza en gran medida en el campo humano, tanto con fines de investigación como de diagnóstico y consiste en NGS - Secuenciación de próxima generación de un único genoma completo, para mapear las mutaciones de nucleótido único (SNP), inserciones y deleciones de más o secuencias menos largas que se han producido en determinadas localizaciones del genoma, y variaciones en el número de copias de porciones / genes genómicos (CNV, variantes de número de copias).
Este procedimiento ayuda a comprender el mecanismo de desarrollo de algunas patologías, con el fin de identificar las instrucciones para un futuro tratamiento clínico como, por ejemplo, en el caso del cáncer. De hecho, mediante este método, el patrimonio genético de un paciente con cáncer puede decodificarse por completo tanto en tejido normal como canceroso, lo que nos permite comprender exactamente qué ha cambiado dentro del genoma y, si es posible, cómo intervenir con medidas específicas.
El procedimiento de nueva secuenciación requiere que el ADN de un individuo se rompa mecánicamente en fragmentos de pequeñas dimensiones (400-500 pares de bases) y que las partes de ADN artificiales denominadas adaptadores se unan a estos fragmentos; Los adaptadores permiten unir los fragmentos de ADN humano a una superficie de vidrio sobre la que se realiza la lectura de bases (A, C, G, T). La lectura de los pares de bases de ADN se realiza mediante reacciones químicas, es decir, la incorporación de nucleótidos que han sido marcados por moléculas fluorescentes. Los millones de secuencias (lecturas) así obtenidas se mapean luego en el genoma de referencia humano mediante un software específico y todas las variantes se identifican comparando el genoma analizado con el genoma de referencia.
Este mismo procedimiento se ha realizado en el genoma humano en Priorix® Tetra lot n. A71CB256A, genoma que pertenece a la línea celular MRC-5 (de origen fetal); el trabajo lo ha llevado a cabo una empresa de EE. UU. que se ocupa habitualmente del análisis de la nueva secuenciación del genoma humano. *
* El nombre del laboratorio que ha realizado el análisis se incluirá en la próxima denuncia formal que presentaremos ante el Fiscal de Roma y también ante los organismos reguladores italianos y europeos. Las asociaciones que están presentando el análisis financiado por Corvelva también se mantendrán al día con estos impactantes resultados. No negamos que nos sentimos, especialmente como padres, angustiados por estos resultados que informamos, como si lo que hemos descubierto hasta ahora no fuera suficiente para preocuparnos.
Resultados
Se encontró que el genoma de referencia humano se correspondía con lecturas del 99.76% del ADN de la vacuna, lo que significa casi en su totalidad. El ADN fetal humano presentado en esta vacuna es un genoma completo único, lo que significa que la vacuna contiene ADN genómico con todos los cromosomas de un individuo masculino (de hecho, el MRC-5 se origina en un feto masculino).
A continuación se presentan los resultados del análisis de diferentes tipos de variantes en comparación con el genoma humano de referencia.
Variante de un solo nucleótido (SNP - polimorfismo de un solo nucleótido) e inserciones / deleciones cortas (InDels)
Las variantes de bases simples de ADN (SNP) son polimorfismos, lo que significa mutaciones de material genético de un solo nucleótido.
Los 'InDels' son, en cambio, pequeñas inserciones y deleciones de menos de 50 pb de longitud y constituyen una clase diferente de variantes genómicas en el genoma humano.
En el genoma humano de la vacuna, se han identificado 3.6 millones de SNP (98.31% de los cuales ya se informaron en la base de datos pública dbSNP y 61.805 nuevos, lo que significa original en este ADN) y alrededor de 804.000 InDels (89.42% de los cuales ya se informaron en dbSNP y 85.106 nuevo).
La cantidad de SNP está en línea con lo que se ha informado en la literatura sobre / en "genoma humano típico", mientras que el InDels da como resultado una cantidad mayor en comparación con lo que ha informado "The 1000 Genomes Project Consortium", es decir, 800.000 en comparación con 600.000 .
CNV (Variantes de número de copia) y SV (Variantes estructurales)
Las variantes del número de copias (CNV) son variantes genómicas debido a las variaciones en el número de copias de fragmentos relativamente grandes (más de 50 pb) entre genomas individuales. Hay dos tipos de CNV: escriba "ganancia" (ganancia de copias) y tipo "pérdida" (pérdida de copias). Se detectaron 218 CNV en el genoma de la vacunación humana, de los cuales 82 fueron "ganancia" (cubriendo una porción del genoma de aproximadamente 6.9 millones de pares de bases) y 136 CNV del tipo "perdida" (cubriendo una porción del genoma de aproximadamente 70 millones de bases).
Según lo descrito por The 1000 Genomes Project Consortium en "Una referencia global para la variación genética humana (Nature, vol. 526, 10 de octubre de 2015)", un genoma humano típico contiene entre 2,100 y 2,500 variantes grandes, que incluyen:
- 1,000 eliminaciones grandes
- 160 variantes en número de copias (CNV)
- 10 inversiones
Que juntas afectan a los 20 millones de bases de la secuencia, considerando también las inserciones.
Como se ve en los INDEL cortos, incluso en el caso de inserciones y deleciones grandes, el genoma de la vacuna no está en línea con un genoma humano "normal", siendo mucho más "reordenado" que el genoma de una persona común.
Visión circular del genoma (trama circos)
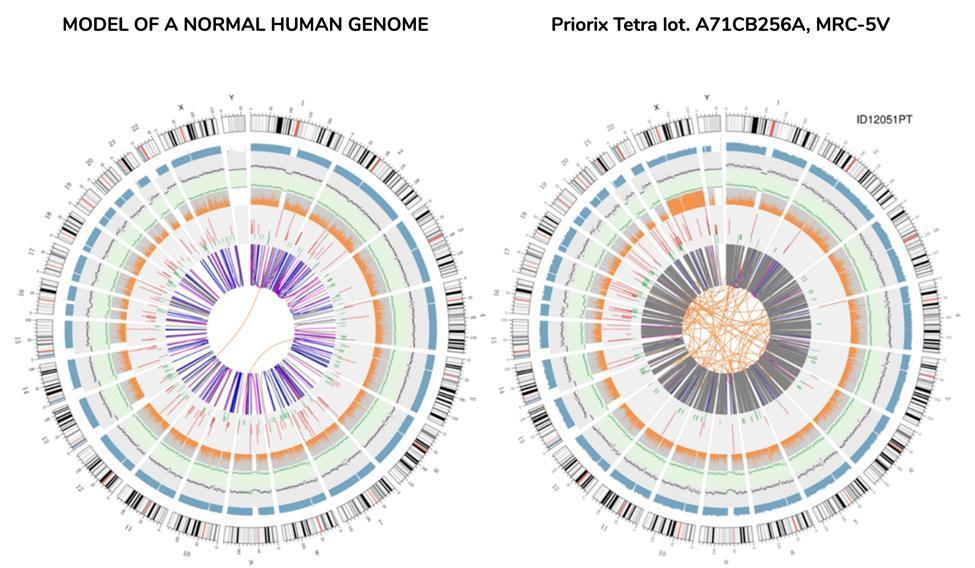
A continuación se muestra una representación gráfica del genoma de la vacuna llamada "diagrama de circos" (comúnmente usado para representar un genoma re-secuenciado), junto con otro que representa un genoma re-secuenciado del ADN extraído de la sangre de un individuo sano - genoma "normal" :
Significado de los círculos concéntricos
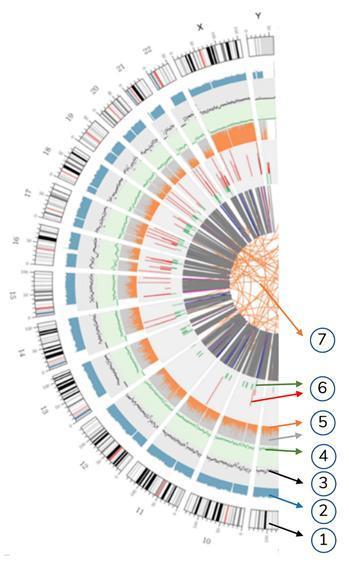
7) El anillo más central representa la inferencia SV (variantes estructurales) en las regiones de exón y empalme. TRA (naranja, translocaciones), INS (verde, inserciones), DEL (eliminaciones, gris), DUP (duplicaciones, rosa) e INV (inversiones, azul).
6) el sexto círculo representa la inferencia CNV (variantes de número de copia). El rojo significa que las secuencias de ADN ganan y el verde significa pérdida.
5) El quinto anillo representa la proporción de SNP en homocigosidad (naranja) y heterocigosidad (gris) en el diseño del histograma.
4) El cuarto anillo (verde) representa la densidad de SNP en el diseño del gráfico de dispersión.
3) El tercer anillo (negro) representa la densidad de INDEL en el diseño del gráfico de dispersión.
2) El segundo anillo (azul claro) representa las lecturas que cubren en el diseño del histograma.
1) El círculo externo (el primer círculo) es el número de cromosoma.
También se puede hacer una comparación aproximada entre el ADN fetal y el ADN de las células HeLa, la línea celular inmortalizada utilizada también para producir la vacuna contra la polio.
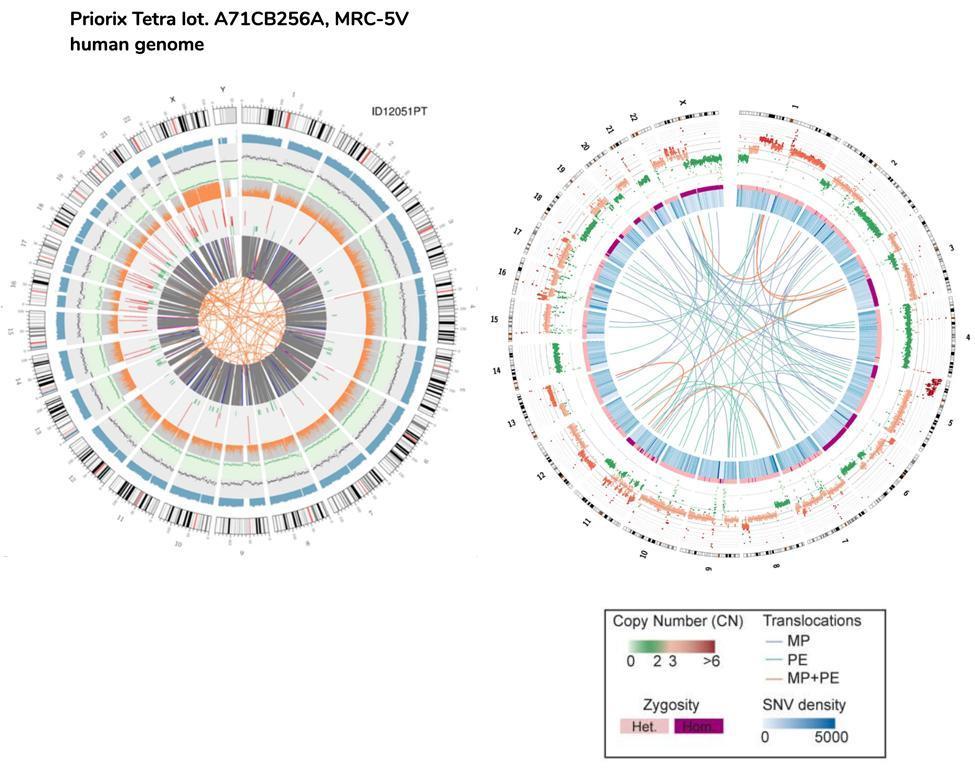
Tenga en cuenta que las translocaciones de células HeLa representadas en la trama de los circos por las líneas del núcleo, se refieren a todo el genoma (de ahí la parte codificante y no codificante), mientras que en el caso de las células de vacunación fetal se refieren solo a genes codificadores .
No hace falta ser un científico para entender de los circos, simplemente de un vistazo, que el genoma de la vacuna no es un genoma que pueda definirse como "normal". Las líneas naranjas entrelazadas en el centro de los circos, no tan numerosas en el anillo correspondiente del genoma "normal", ya dan sentido a la anomalía de este genoma.
Análisis de variantes en genes cancerosos
El análisis de las variantes SNP, InDels, CNV, SV en 560 genes elegidos porque participan en diferentes formas de cáncer humano muestra la presencia de numerosas variantes "originales", es decir, que ni siquiera están presentes en las bases de datos públicas, por lo tanto están no conocido en la literatura. En otras palabras, se han identificado modificaciones importantes de genes que se sabe que están asociados con diversas formas tumorales, para todos los 560 genes verificados; Además, hay variantes cuyas consecuencias no se conocen, pero que, sin embargo, afectan a los genes implicados en la inducción del cáncer humano.
Conclusión
El ADN genómico humano contenido en la vacuna del lote Priorix. n. A71CB256A es evidentemente anómalo, presentando inconsistencias importantes si se compara con un genoma humano típico, es decir, el de un ser humano sano. Existen varias variantes desconocidas (que no se mencionan en las bases de datos públicas) y algunas de ellas se encuentran en genes involucrados en el cáncer. Lo que también es aparentemente anómalo es el exceso de genoma que muestra cambios en el número de copias (CNV) y variantes estructurales (SV), como translocaciones, inserciones, deleciones, duplicaciones e inversiones, muchas de las cuales involucran genes.
Se desconoce la contribución potencial de las numerosas variantes (no presentes en la literatura científica y en las bases de datos públicas) al fenotipo de las células utilizadas para el crecimiento de los virus de la vacuna.
Dentro del PDF encontrará todas las ideas relacionadas también con las implicaciones para el saludo y la correspondencia con la EMA.
Descargar: CORVELVA-MRC-5-contenía-in-Priorix-Tetra-Completa-de secuenciación del genoma