Vaccinegate: Sequenciação completa do genoma de MRC-5 contida no Priorix Tetra

Estas últimas análises foram possíveis graças à contribuição ativa das associações francesas Associação Liberté Informações Santé (ALIS), Liga nacional para a libertação de vacinas (LNPLV) e a associação australiana Rede Australiana de Riscos de Vacinação (AVN) que agradecemos.
Os sequenciadores de nova geração tornaram-se instrumentos de escolha para análises aprofundadas no campo da biologia e da medicina, especialmente o de precisão. Essas ferramentas permitem uma abordagem nova e mais global para uma série de aplicações, como sequenciamento de novo, estudos de metagenômica, epigenômica, sequenciamento de transcriptoma e sequenciamento de genoma.
Essa última aplicação (re-sequenciamento) é difundida no campo humano, tanto para fins de pesquisa quanto de diagnóstico, e consiste no sequenciamento com a tecnologia NGS (Next Generation Sequencing) de um genoma individual inteiro para mapear mutações de nucleotídeo único (SNP, pronúncia 'snip'), inserções e deleções de sequências mais ou menos longas ocorreram em certas posições do genoma e variações no número de cópias de partes de genoma / genes (CNV, Copy Number Variants).
Este procedimento é útil para entender os mecanismos de desenvolvimento de algumas patologias, a fim de identificar as direções para o futuro tratamento clínico, como no caso do câncer. Com esse método, de fato, o patrimônio genético de um paciente com câncer pode ser completamente decodificado no tecido normal e tumoral, permitindo entender o que mudou no genoma e, se possível, intervir nos protocolos direcionados.
O procedimento de re-sequenciamento requer que o DNA de um indivíduo seja mecanicamente quebrado em pequenos fragmentos (400-500 pares de bases) e que os fragmentos estejam vinculados a tratos artificiais de DNA chamados adaptadores, que permitem ligar os fragmentos de DNA humano para uma superfície de vidro na qual as bases são então lidas (A, C, G, T). As bases do DNA são lidas incorporando reações químicas de nucleotídeos marcados com moléculas fluorescentes. Os milhões de sequências (leituras) obtidas a partir do sequenciamento na superfície do vidro são então mapeadas no genoma de referência humano com software apropriado e, portanto, todas as variantes presentes no genoma analisado são identificadas em relação à referência.
Este mesmo procedimento foi realizado no genoma humano presente no lote Priorix Tetra. n. A71CB256A, genoma pertencente à linha celular MRC-5 (de origem fetal); o trabalho foi realizado em uma empresa localizada nos EUA, que lida rotineiramente com a análise de genomas humanos por sequenciamento. *
* O nome do laboratório que realizou essa análise será incluído na próxima declaração que depositaremos no Ministério Público em Roma, bem como nos órgãos de controle italiano e europeu. As realidades que estão depositando os resultados das análises financiadas pela Corvelva também serão imediatamente atualizadas com esses resultados intrigantes. Não negamos que, como pais em primeiro lugar, estamos angustiados com os resultados que relatamos abaixo - se o que descobrimos até agora não foi suficiente.
Resultados
Verificou-se que o genoma de referência humano é coberto por leituras originárias do DNA da vacina para 99.76%, portanto, para quase toda a sua totalidade. O DNA humano fetal representado nesta vacina é, portanto, um genoma individual completo ou está presente na vacina de DNA genômico de todos os cromossomos de um indivíduo do sexo masculino (e de fato o feto do qual a linha celular MRC-5 deriva é masculino).
Abaixo estão os resultados da análise dos vários tipos de variantes com relação ao genoma de referência humano.
Variantes de nucleotídeo único (SNP) e inserções / deleções curtas (InDels)
As variantes de bases individuais de DNA (SNP, pronunciado "snip") são polimorfismos, ou seja, variações do material genético, carregadas por um único nucleotídeo. Os 'InDels', por outro lado, são pequenas inserções e deleções com menos de 50 pb de comprimento e constituem outra classe de variantes genômicas no genoma humano.
Um total de aproximadamente 3.6 milhões de SNPs foram identificados no genoma da vacina humana (dos quais 98.31% já relatados no banco de dados público dbSNP e 61.805 novos ou originais desse DNA) e aproximadamente 804 mil InDels (dos quais 89.42% já relatados no dbSNP e 85.106 novos).
A quantidade de SNP está alinhada com a relatada na literatura em um "genoma humano típico", enquanto os indels estão em uma quantidade maior do que a relatada pelo "The 1000 Genomes Project Consortium" ou 800 mil em comparação a 600 mil.
CNV (variantes do número de cópias) e SV (variantes estruturais)
Variantes no número de cópias (CNV) são variantes genômicas devido a variações no número de cópias de fragmentos relativamente grandes (maiores que 50 pb) entre os genomas individuais. Existem dois tipos de CNV: tipo "ganho" (ganho de cópias) e tipo "perda" (perda de cópias). 218 CNVs foram detectados no genoma da vacina humana, 82 dos quais são do tipo "ganho" (cobrindo uma parte do genoma igual a aproximadamente 6.9 milhões de pares de bases) e 136 "CNVs" do tipo "perda" (que cobrem uma parte do genoma cerca de 70 milhões de bases).
Conforme descrito pelo The 1000 Genomes Project Consortium em "Uma referência global para variação genética humana (Nature, vol. 526, 10 de outubro de 2015)", um genoma humano típico contém de 2.100 a 2.500 grandes variantes, incluindo:
- 1.000 exclusões grandes
- 160 variações no número de cópias (CNVs)
- 10 reversões
que influenciam globalmente, considerando também as inserções, em 20 milhões de bases de sequência.
Como observado para INDELs curtos, mesmo no caso de grandes inserções e deleções, o genoma da vacina não está alinhado com um genoma humano 'normal', sendo muito mais "reorganizado" do que o genoma de um indivíduo comum.
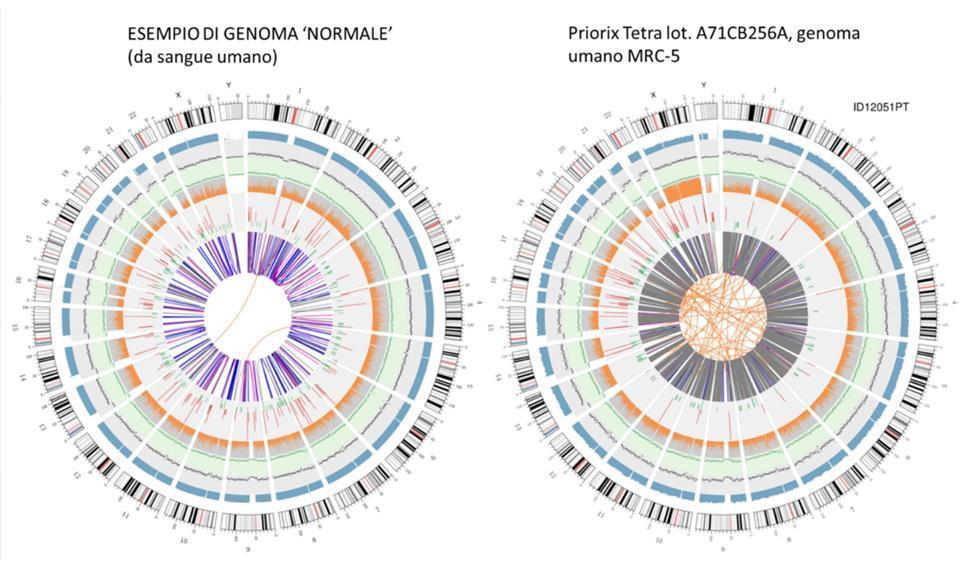
Visualização circular do genoma (circos plot)
Uma representação gráfica do genoma da vacina chamada "circos plot" (que é comumente usada para representar um genoma re-sequenciado) é mostrada abaixo, ao lado de outra que representa um genoma re-sequenciado a partir do DNA extraído do sangue de um indivíduo saudável - genoma "normal":
Significado dos vários círculos concêntricos
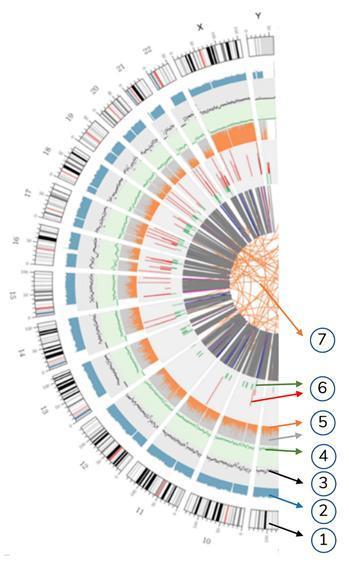
7) O anel mais central representa a inferência de SV (variantes estruturais) nas regiões exônicas e de união. ENTRE (laranja, translocações), INS (verde, inserções), DEL (exclusões, cinza), DUP (duplicações, rosa) e INV (inversões, azul).
6) O sexto toque representa a inferência da CNV (variantes no número de cópias). Vermelho significa ganhar pedaços de DNA e verde significa perda.
5) O quinto anel representa a proporção de SNP na homozigose (laranja) e na heterozigose (cinza) no estilo do histograma.
4) O quarto anel (verde) representa o snp de densidade no estilo "gráfico de dispersão".
3) O terceiro anel (preto) representa a densidade dos INDELs no estilo "gráfico de dispersão".
2) O segundo toque (azul) representa a cobertura das leituras no estilo do histograma.
1) O círculo externo (o primeiro círculo) é o número do cromossomo.
Também pode ser feita uma comparação aproximada entre o DNA fetal e o DNA celular HeLa, a linha celular imortalizada também usada na produção da vacina contra a poliomielite.
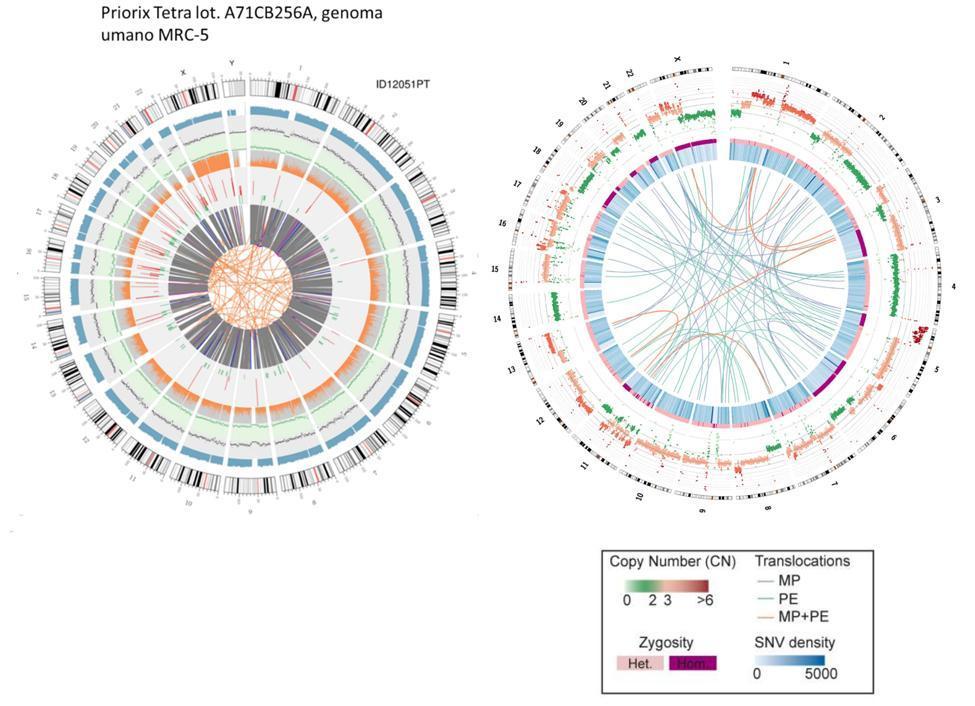
Deve-se enfatizar que as translocações das células HeLa representadas no gráfico de circos pelas linhas do núcleo se referem a todo o genoma (portanto parte codificante e não codificante), enquanto que no caso das células da vacina fetal se referem apenas aos genes codificadores.
Não é preciso ser cientista para entender desde os circos, simplesmente de relance, que o genoma da vacina não é um genoma que se possa definir como "normal". As linhas laranja entrelaçadas no centro dos circos, não tão numerosas no anel correspondente do genoma "normal", já nos fazem adivinhar a anomalia desse genoma.
conclusões
O DNA genômico humano contido na vacina lote Prorix. n. O A71CB256A é evidentemente anômalo, apresentando inconsistências importantes em relação a um genoma humano típico, ou seja, de um indivíduo saudável. Existem inúmeras variantes desconhecidas (não registradas em bancos de dados públicos) e várias delas estão localizadas em genes envolvidos no câncer. O que também é evidentemente anômalo é o excesso de genoma, que mostra alterações no número de cópias (CNV) e variantes estruturais (SV), como translocações, inserções, deleções, duplicações e inversões, muitas das quais envolvendo genes.
A contribuição potencial de muitas variantes (não presentes na literatura científica e em bancos de dados públicos) para o fenótipo das células utilizadas para o crescimento de vírus vacinais é desconhecida.
Dentro do PDF, você encontrará todos os insights também relacionados às implicações para a saúde e a correspondência com a EMA.
Download: CORVELVA-Ri-genoma-sequenciação-humano-Priorix-Tetra.pdf