Vaccinegate: MRC-5 contained in Priorix Tetra - Complete genome sequencing

These latest analyzes were made possible thanks to the active contribution of the French associations Association Liberté Informations Santé (ALIS), National League for Freedom of Vaccinations (LNPLV) and the Australian Association Australian Vaccination-risks Network (AVN), that we thank.
New generation sequencing have become the preferred tool for in-depth analysis in the field of biology and medical science, especially high precision ones. Thanks to these tools, we can approach in a more modern and comprehensive way a number of applications such as de novo sequencing, metagenomic and epigenomic studies, transcriptome sequencing and genome re-sequencing.
This last one (re-sequencing) is largely used in human field, both for research and diagnostic purposes and consists of NGS - Next Generation Sequencing of an entire single genome, to map the Single Nucleotide mutations (SNP), insertions and deletions of more or less long sequences that have occurred in certain locations of the genome, and variations in the number of copies of genomic portions / genes (CNV, Copy Number Variants).
This procedure helps to understand the development mechanism of some pathologies, in order to identify the directions for a future clinical treatment as in the case of cancer for example. Indeed, by this method the genetic heritage of a cancer patient can be fully decoded in both normal and cancerous tissue, thus allowing us to comprehend what exactly has changed within the genome, and, if possible, how to intervene with targeted measures.
The re-sequencing procedure requires that the DNA of an individual is mechanically broken into small dimension fragments (400-500 base pairs) and artificial DNA parts named adapters are tied to these fragments; adapters make it possible to tie the human DNA fragments to a glass surface on which the bases reading (A, C, G, T) is performed. The DNA base pairs reading takes place by means of chemical reactions, namely the incorporation of nucleotides that have been marked by fluorescent molecules. The million sequences (reads) thus obtained are then mapped on the human reference genome by specific software and all the variants are identified comparing the analyzed genome with the reference genome.
This same procedure has been performed on the human genome in Priorix® Tetra lot n. A71CB256A, genome which belongs to cell line MRC-5 (of fetal origin); the work has been carried out by a company in the USA, that routinely deals with human genome re-sequencing analysis. *
* the name of the laboratory that has performed the analysis will be included in the next formal complaint we will file at the Public Prosecutor of Rome and as well at the Italian and European regulatory bodies. The associations who are filing the analysis funded by Corvelva will be promptly kept up to date with these shocking results too. We are no denying that we feel, especially as parents, distressed by these results we are reporting - as if what we have found out so far was not enough to worry about.
Results
The human reference genome was found to be matched by 99.76% reads from vaccine DNA, which means nearly in all its entirety. The human fetal DNA presented in this vaccine is a single entire genome, that means the vaccine contains genomic DNA with all the chromosomes of a male individual (in fact MRC-5 originates from a male fetus).
Given below are the analysis results of different types of variants compared to the reference human genome.
Single nucleotide variant (SNP - single nucleotide polymorphism) and short insertions / deletions (InDels)
DNA single base variants (SNP) are polymorphisms, which means genetic material mutations of a single nucleotide.
The 'InDels' are instead small insertions and deletions of less than 50 bp length and constitute a different class of genomic variants in the human genome.
In the vaccine human genome, 3.6 million SNP have been identified (98.31% of which are already reported in the public database dbSNP and 61.805 new, that means original in this DNA) and about 804.000 InDels (89.42% of which already reported in dbSNP and 85.106 new).
The amount of SNP is in line with what has been reported in literature on / in "typical human genome", whereas the InDels results in a higher quantity compared to what has been reported by "The 1000 Genomes Project Consortium" namely 800.000 compared to 600.000 .
CNV (Copy Number Variants) and SV (Structural Variants)
The copy number variants (CNVs) are genomic variants due to variations in the number of copies of relatively large fragments (longer than 50 bp) between individual genomes. There are two types of CNVs: type "gain" (gain of copies) and type "loss" (loss of copies). 218 CNVs were detected in the human vaccination genome, of which 82 were "gain" (covering a portion of the genome of about 6.9 million base pairs) and 136 CNVs of the "loss" type (covering a portion of the genome of about 70 million bases).
As described by The 1000 Genomes Project Consortium in "A global reference for human genetic variation (Nature, vol. 526, 10 Oct. 2015)" a typical human genome contains from 2,100 to 2,500 large variants including:
- 1,000 large deletions
- 160 variants in number of copies (CNVs)
- 10 investments
Which together affect the 20 million bases of the sequence, considering the insertions as well.
As seen for the short INDELs, even in the case of large insertions and deletions, the vaccine genome is therefore not in line with a "normal" human genome, being much more "rearranged" than a genome of a common person.
Circular vision of genome (circus plot)
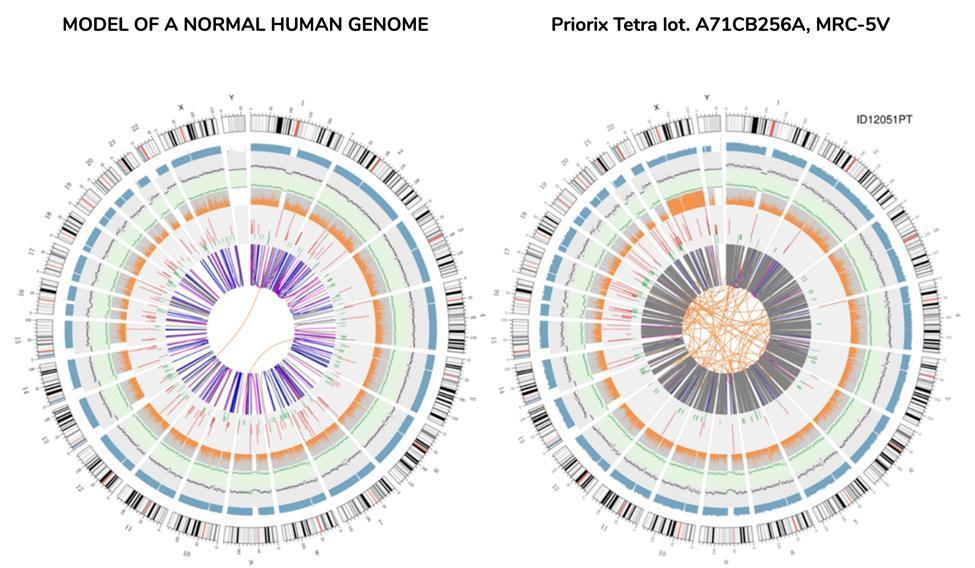
A graphical representation of the vaccine genome called "circos plot" (commonly used to represent a re-sequenced genome), is shown below, alongside another representing a genome re-sequenced from DNA extracted from blood of a healthy individual - "normal" genome :
Meaning of the concentric circles
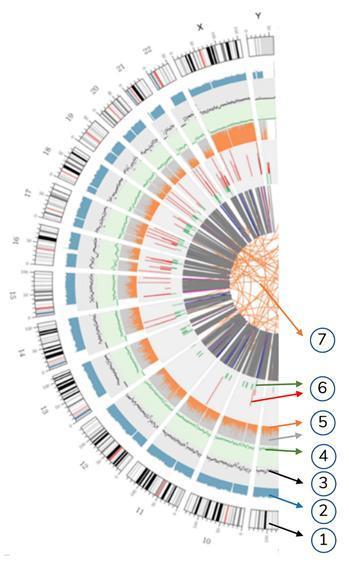
7) The most central ring represents the SV (Structural Variants) inference in the exon and splicing regions. BETWEEN (orange, translocations), INS (green, insertions), DEL (deletions, gray), DUP (duplications, pink) and INV (inversions, blue).
6) the sixth circle represents the CNV inference (copy number variants). Red means DNA sequences gain and green means loss.
5) The fifth ring represents the SNP proportion in homozygosity (orange) and heterozygosity (gray) in histogram layout.
4) The fourth ring (green) represents the SNP density in scatter chart layout.
3) The third ring (black) represents the INDELs density in scatter chart layout.
2) The second ring (light blue) represents the reads covering in histogram layout.
1) The external circle (the first circle) is the chromosome number.
An approximate comparison can also be made between fetal DNA and the HeLa cells DNA, the immortalized cell line used as well to produce the polio vaccine.
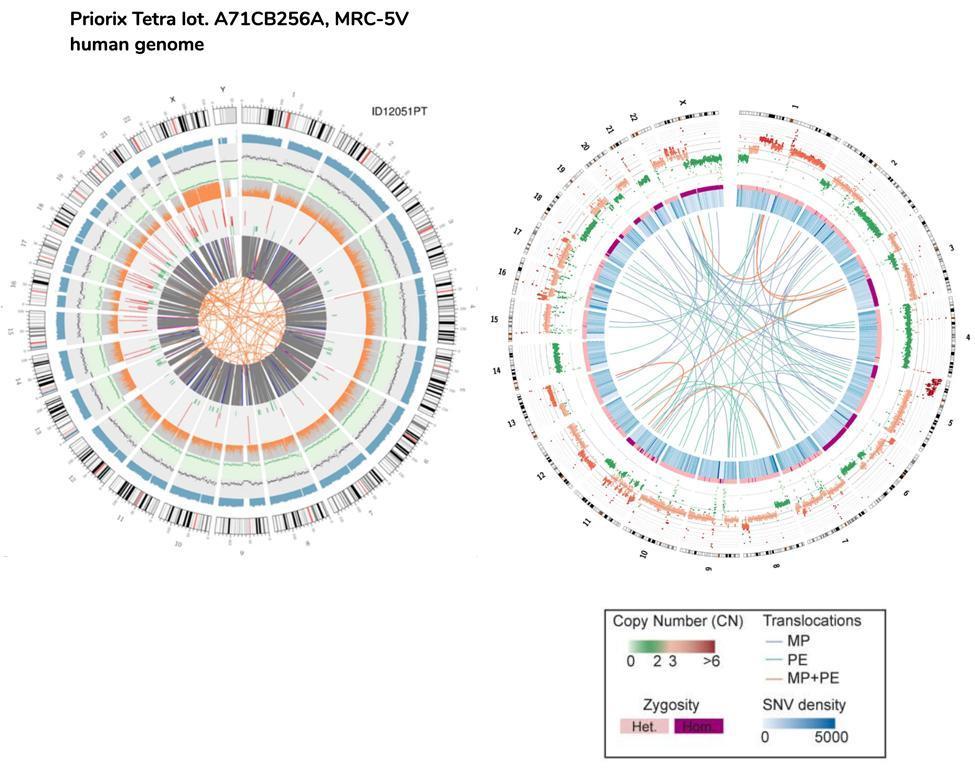
Please note that the HeLa cells translocations represented in the circos plot by the lines of the nucleus, are referred to the entire genome (hence the coding and non-coding part), while in the case of fetal vaccination cells they refer only to coding genes .
There is no need to be a scientist to understand from the circos, simply at a glance, that the vaccine genome is not a genome that can be defined as "normal". The orange lines intertwined at the center of the circos, not so numerous in the corresponding ring of the "normal" genome, already make sense to the anomaly of this genome.
Variant analysis in cancer genes
The analysis on SNP, InDels, CNV, SV variants on 560 genes chosen because they involved in different forms of human cancer shows the presence of numerous "original" variants, that's to say that they are, not even present in public databases, therefore are not known in the literature. In other words, important modifications of genes known to be associated with various tumor forms have been identified, for all the 560 verified genes; furthermore, there are variants whose consequences are not known, but which, however, affects genes involved in the induction of human cancer.
Conclusion
The human genomic DNA contained in the Priorix lot vaccine. n. A71CB256A is evidently anomalous, presenting important inconsistencies if compared to a typical human genome, ie the one of a healthy human being. There are several unknown variants (not noted in public databases) and some of them are located in genes involved in cancer. What is also apparently anomalous, is the excess of genome that shows changes in the number of copies (CNV) and structural variants (SV), such as translocations, insertions, deletions, duplications and inversions, many of which involve genes.
The potential contribution of the numerous variants (not present in the scientific literature and in public databases) to the phenotype of the cells used for the growth of vaccine viruses is not known.
Inside the PDF you will find all the insights related also to the implications for the greeting and the correspondence with the EMA.
Download: CORVELVA-MRC-5-contained-in-Priorix-Tetra-Complete-genome-sequencing