Vaccinegate: MRC-5 in Priorix Tetra - Vollständige Genomsequenzierung

Diese neuesten Analysen wurden dank des aktiven Beitrags der französischen Verbände ermöglicht Verein Liberté Informationen Santé (ALIS), Ligue nationale pour la liberté des Impfungen (LNPLV) und der australischen Vereinigung Australisches Netzwerk für Impfrisiken (AVN), das danken wir.
Die Sequenzierung der neuen Generation ist zum bevorzugten Werkzeug für eingehende Analysen auf dem Gebiet der Biologie und Medizin geworden, insbesondere für hochpräzise. Dank dieser Tools können wir eine Reihe von Anwendungen wie De-novo-Sequenzierung, metagenomische und epigenomische Studien, Transkriptomsequenzierung und Genom-Re-Sequenzierung auf modernere und umfassendere Weise angehen.
Diese letzte (Re-Sequenzierung) wird hauptsächlich für Forschungs- und Diagnosezwecke im menschlichen Bereich verwendet und besteht aus NGS - Next Generation Sequencing eines gesamten einzelnen Genoms, um die Single Nucleotide Mutationen (SNP), Insertionen und Deletionen von mehr abzubilden oder weniger lange Sequenzen, die an bestimmten Stellen des Genoms aufgetreten sind, und Variationen in der Anzahl der Kopien von genomischen Teilen / Genen (CNV, Kopienzahlvarianten).
Dieses Verfahren hilft, den Entwicklungsmechanismus einiger Pathologien zu verstehen, um die Richtungen für eine zukünftige klinische Behandlung wie beispielsweise bei Krebs zu identifizieren. In der Tat kann mit dieser Methode das genetische Erbe eines Krebspatienten sowohl im normalen als auch im krebsartigen Gewebe vollständig entschlüsselt werden, sodass wir nachvollziehen können, was sich im Genom genau verändert hat und wie wir, wenn möglich, mit gezielten Maßnahmen eingreifen können.
Das Re-Sequenzierungsverfahren erfordert, dass die DNA eines Individuums mechanisch in Fragmente kleiner Dimension (400-500 Basenpaare) zerlegt wird und künstliche DNA-Teile, die als Adapter bezeichnet werden, an diese Fragmente gebunden werden; Adapter ermöglichen es, die menschlichen DNA-Fragmente an eine Glasoberfläche zu binden, auf der die Basenablesung (A, C, G, T) durchgeführt wird. Das Lesen der DNA-Basenpaare erfolgt mittels chemischer Reaktionen, nämlich des Einbaus von Nukleotiden, die durch fluoreszierende Moleküle markiert wurden. Die so erhaltenen Millionen Sequenzen (Reads) werden dann durch spezifische Software auf das menschliche Referenzgenom abgebildet und alle Varianten werden identifiziert, indem das analysierte Genom mit dem Referenzgenom verglichen wird.
Das gleiche Verfahren wurde am menschlichen Genom in Priorix® Tetra Los n durchgeführt. A71CB256A, Genom, das zur Zelllinie MRC-5 (fötalen Ursprungs) gehört; Die Arbeit wurde von einem Unternehmen in den USA durchgeführt, das sich routinemäßig mit der Analyse der Sequenzierung des menschlichen Genoms befasst. * *
* Der Name des Labors, das die Analyse durchgeführt hat, wird in die nächste formelle Beschwerde aufgenommen, die wir bei der Staatsanwaltschaft von Rom sowie bei den italienischen und europäischen Regulierungsbehörden einreichen werden. Die Verbände, die die von Corvelva finanzierte Analyse einreichen, werden auch mit diesen schockierenden Ergebnissen umgehend auf dem Laufenden gehalten. Wir leugnen nicht, dass wir uns besonders als Eltern von diesen Ergebnissen, über die wir berichten, beunruhigt fühlen - als ob das, was wir bisher herausgefunden haben, nicht genug wäre, um uns Sorgen zu machen.
Die Ergebnisse
Es wurde festgestellt, dass das humane Referenzgenom zu 99.76% mit der DNA des Impfstoffs übereinstimmt, dh nahezu vollständig. Die in diesem Impfstoff vorgestellte humane fetale DNA ist ein einzelnes Gesamtgenom, dh der Impfstoff enthält genomische DNA mit allen Chromosomen eines männlichen Individuums (MRC-5 stammt tatsächlich von einem männlichen Fötus).
Nachstehend sind die Analyseergebnisse verschiedener Arten von Varianten im Vergleich zum menschlichen Referenzgenom angegeben.
Einzelnukleotidvariante (SNP - single nucleotide polymorphism) und kurze Insertionen / Deletionen (InDels)
DNA-Single-Basen-Varianten (SNP) sind Polymorphismen, dh Mutationen des genetischen Materials eines einzelnen Nukleotids.
Die "InDels" sind stattdessen kleine Insertionen und Deletionen von weniger als 50 bp Länge und bilden eine andere Klasse genomischer Varianten im menschlichen Genom.
Im menschlichen Genom des Impfstoffs wurden 3.6 Millionen SNP (98.31% davon bereits in der öffentlichen Datenbank dbSNP und 61.805 neue, dh in dieser DNA ursprüngliche) und etwa 804.000 InDels (89.42% davon bereits in dbSNP und gemeldet) identifiziert 85.106 neu).
Die Menge an SNP entspricht den Angaben in der Literatur zu / im "typischen menschlichen Genom", während die InDels zu einer höheren Menge führen als die Angaben des "The 1000 Genomes Project Consortium", nämlich 800.000 im Vergleich zu 600.000 .
CNV (Copy Number Variants) und SV (Structural Variants)
Die Kopienzahlvarianten (CNVs) sind genomische Varianten aufgrund von Variationen in der Kopienzahl relativ großer Fragmente (länger als 50 bp) zwischen einzelnen Genomen. Es gibt zwei Arten von CNVs: Typ "Gewinn" (Gewinn von Kopien) und Typ "Verlust" (Verlust von Kopien). Im menschlichen Impfgenom wurden 218 CNVs nachgewiesen, von denen 82 "Gain" (einen Teil des Genoms von etwa 6.9 Millionen Basenpaaren abdeckend) und 136 CNVs vom "Loss" -Typ (einen Teil des Genoms von etwa 70 abdeckend) waren Millionen Basen).
Wie vom 1000 Genomes Project Consortium in "Eine globale Referenz für die genetische Variation des Menschen (Nature, Bd. 526, 10. Oktober 2015)" beschrieben, enthält ein typisches menschliches Genom 2,100 bis 2,500 große Varianten, darunter:
- 1,000 große Deletionen
- 160 Varianten in Anzahl der Exemplare (CNVs)
- 10 Inversionen
Was zusammen die 20 Millionen Basen der Sequenz betrifft, auch unter Berücksichtigung der Insertionen.
Wie für die kurzen INDELs zu sehen ist, stimmt das Impfgenom selbst bei großen Insertionen und Deletionen nicht mit einem "normalen" menschlichen Genom überein, da es viel "umgeordneter" ist als das Genom einer gewöhnlichen Person.
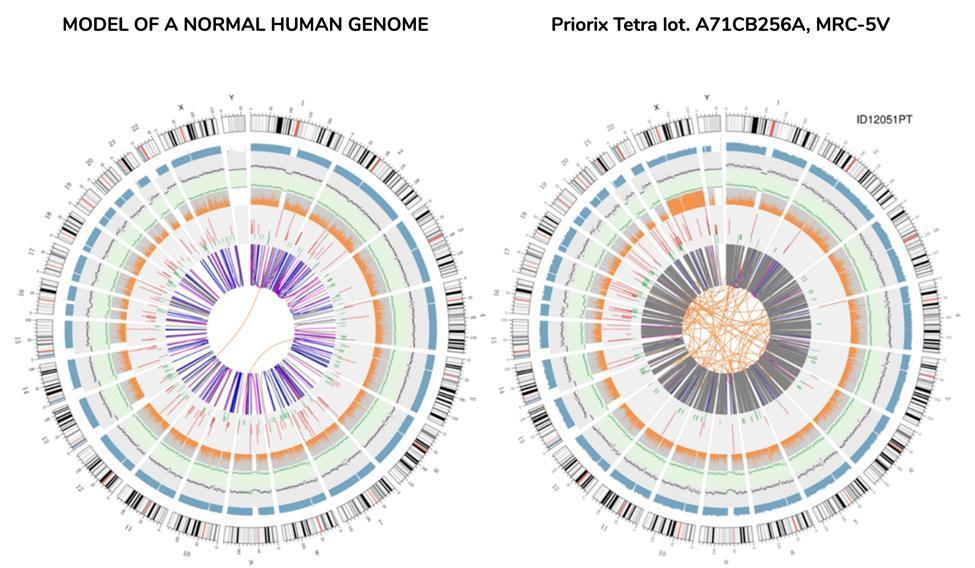
Zirkuläre Vision des Genoms (Circos Plot)
Eine grafische Darstellung des Impfstoffgenoms mit der Bezeichnung "circos plot" (üblicherweise zur Darstellung eines neu sequenzierten Genoms verwendet) ist nachstehend neben einem anderen Genom dargestellt, das aus DNA-Sequenzen aus dem Blut eines gesunden Individuums - dem "normalen" Genom - neu sequenziert wurde :
Bedeutung der konzentrischen Kreise
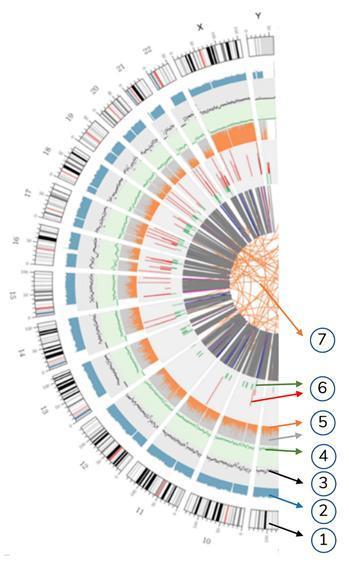
7) Der zentralste Ring repräsentiert die SV-Inferenz (Strukturvarianten) in den Exon- und Spleißbereichen. TRA (orange, translocations), INS (grün, insertions), DEL (deletions, grau), DUP (duplications, pink) und INV (inversions, blau).
6) Der sechste Kreis repräsentiert die CNV-Folgerung (Kopienzahlvarianten). Rot bedeutet DNA-Sequenzgewinn und Grün bedeutet Verlust.
5) Der fünfte Ring repräsentiert den SNP-Anteil an Homozygotie (orange) und Heterozygotie (grau) im Histogramm-Layout.
4) Der vierte Ring (grün) repräsentiert die SNP-Dichte im Streudiagramm-Layout.
3) Der dritte Ring (schwarz) repräsentiert die INDEL-Dichte im Streudiagramm-Layout.
2) Der zweite Ring (hellblau) repräsentiert die Anzeige im Histogrammlayout.
1) Der äußere Kreis (der erste Kreis) ist die Chromosomenzahl.
Ein ungefährer Vergleich kann auch zwischen der DNA des Fötus und der DNA der HeLa-Zellen, der immortalisierten Zelllinie, die auch zur Herstellung des Polio-Impfstoffs verwendet wird, durchgeführt werden.
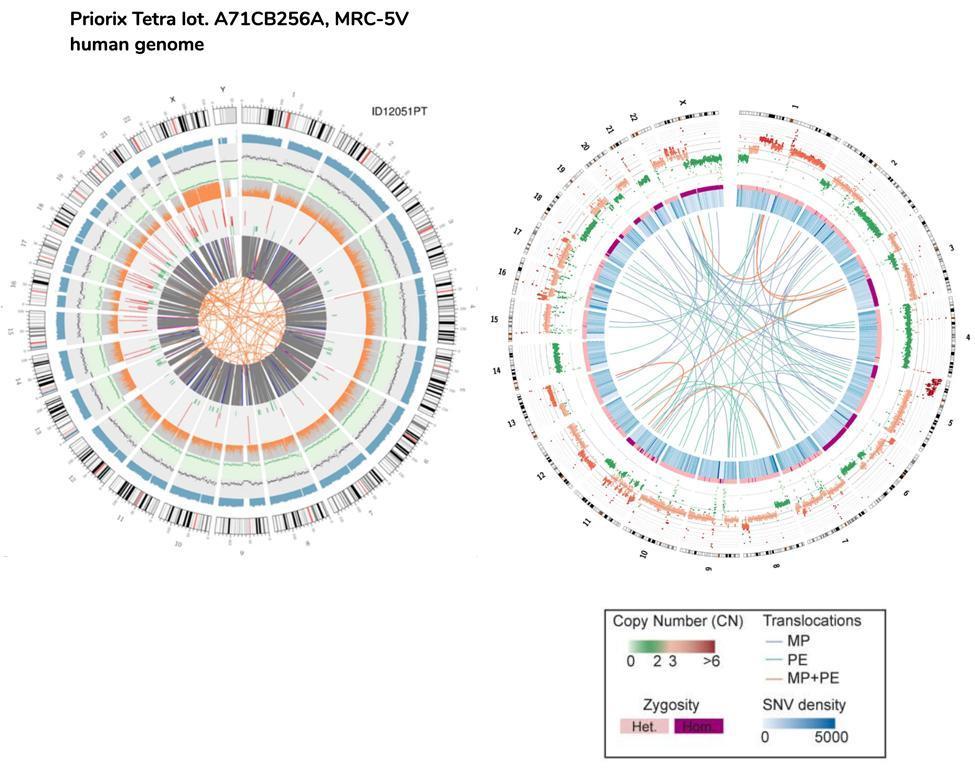
Bitte beachten Sie, dass die HeLa-Zell-Translokationen, die im Circos-Plot durch die Linien des Zellkerns dargestellt sind, sich auf das gesamte Genom beziehen (daher der kodierende und der nicht-kodierende Teil), während sie sich im Fall von fetalen Impfzellen nur auf kodierende Gene beziehen .
Es ist nicht nötig, einen Wissenschaftler zu haben, um aus den Zirkussen auf einen Blick zu verstehen, dass das Impfstoffgenom kein Genom ist, das als "normal" definiert werden kann. Die orangefarbenen Linien in der Mitte des Zirkos, die im entsprechenden Ring des "normalen" Genoms nicht so zahlreich sind, machen für die Anomalie dieses Genoms bereits Sinn.
Variantenanalyse in Krebsgenen
Die Analyse der SNP-, InDels-, CNV- und SV-Varianten von 560 Genen, die ausgewählt wurden, weil sie an verschiedenen Formen von Krebs beim Menschen beteiligt waren, zeigt das Vorhandensein zahlreicher "ursprünglicher" Varianten, das heißt, sie sind, nicht einmal in öffentlichen Datenbanken vorhanden, also vorhanden in der Literatur nicht bekannt. Mit anderen Worten, wichtige Modifikationen von Genen, von denen bekannt ist, dass sie mit verschiedenen Tumorformen assoziiert sind, wurden für alle 560 verifizierten Gene identifiziert; Darüber hinaus gibt es Varianten, deren Folgen nicht bekannt sind, die jedoch Gene betreffen, die an der Auslösung von Krebs beim Menschen beteiligt sind.
Zusammenfassung
Die im Priorix-Chargen-Impfstoff enthaltene humane genomische DNA. n. A71CB256A ist offensichtlich anomal und weist im Vergleich zu einem typischen menschlichen Genom, dh dem eines gesunden Menschen, erhebliche Inkonsistenzen auf. Es gibt mehrere unbekannte Varianten (nicht in öffentlichen Datenbanken vermerkt) und einige von ihnen befinden sich in Genen, die an Krebs beteiligt sind. Anomal erscheint auch der Genomüberschuss, der Veränderungen in der Anzahl der Kopien (CNV) und Strukturvarianten (SV) wie Translokationen, Insertionen, Deletionen, Duplikationen und Inversionen aufweist, von denen viele Gene betreffen.
Der mögliche Beitrag der zahlreichen (in der wissenschaftlichen Literatur und in öffentlichen Datenbanken nicht vorhandenen) Varianten zum Phänotyp der für das Wachstum von Impfviren verwendeten Zellen ist nicht bekannt.
Im PDF finden Sie alle Erkenntnisse, die sich auch auf die Auswirkungen auf die Begrüßung und die Korrespondenz mit der EMA beziehen.
Herunterladen: CORVELVA-MRC-5-contained-in-Priorix-Tetra-Complete-Genom-Sequenzierung