Odpowiedź techniczna na krytykę dr Bucci

Poniżej przedstawiamy odpowiedź techniczną dr Loretty Bolgan na krytykę opublikowaną przez dr Bucci dotyczącą analiz Corvelva.
Drogi Dr Bucci,
poniżej odpowiadam na krytyczne problemy, które poruszyłeś w szczególności w swoim artykule opublikowanym na: BadScientists.com
Przede wszystkim chciałbym zwrócić uwagę, że użycie technologii NGS / HTS (sekwencjonowanie nowej generacji lub sekwencjonowanie wysokiej przepustowości) NIE jest oryginalne na złożonych podłożach pochodzenia biotechnologicznego, takich jak szczepionki.
Technologia NGS została już zastosowana w produktach szczepionkowych, na przykład:
- w analizie komercyjnych partii szczepionki przeciw rotawirusom (Victoria JG, Wang C, Jones MS, Jaing C, McLoughlin K, Gardner S i in. Wirusowe kwasy nukleinowe w żywych atenuowanych szczepionkach: wykrywanie wariantów mniejszościowych i przypadkowego wirusa. J Virol 2010; 84: 6033–40) umożliwiając późniejsze blokowanie wprowadzania do obrotu partii zanieczyszczonych cirkowirusem świń typu 1 (PCV1), pomimo wcześniejszego przejścia wszystkich testów wymaganych do rozwoju, badań klinicznych i produkcji (Dubin, G; Toussaint, JF; Cassart, JP; Howe, B.; Boyce, D.; Friedland, L., Abu-Elyazeed, R., Poncelet, S., Han, HH; Debrus, S. Badanie dochodzenia agencji regulacyjnej w sprawie potencjalnego typu cirkowirusa świń 1 zanieczyszczenie szczepionką przeciwko ludzkiemu rotawirusowi, Rotarix: podejście i wynik Hum. Vaccines Immunother. 2013, 9, 2398–2408).
- w wykrywaniu nowego rabdowirusa w linii komórkowej Sf9 (MH, Galvin TA, Glasner DR, Shaheduzzaman S, Khan AS. Identyfikacja nowego rabdowirusa w liniach komórkowych Spodoptera frugiperda. J Virol 2014; 88: 6576–85)
- w identyfikacji wirusa szczepionki przeciw śwince w mózgu dziecka z obniżoną odpornością, które zmarło na zapalenie mózgu (Morfopoulou S, Mee ET, Connaughton SM, Brown JR, Gilmour K, Chong WK, Duprex WP, Ferguson D, Hubank M, Hutchinson C, Kaliakatsos M, McQuaid S, Paine S, Plagnol V, Ruis C, Virasami A, Zhan H, Jacques TS, Schepelmann S, Qasim W, Breuer J. Głębokie sekwencjonowanie ujawnia obecność wirusa szczepionki przeciw śwince związanego z przewlekłym zapaleniem mózgu Acta Neuropathol. 2017 Jan; 133 (1): 139-147. Doi: 10.1007 / s00401- 016-1629-y. Epub 2016 21 października. PubMed PMID: 27770235).
Również we wspólnej pracy z 2017 r. „A Multicenter Study To Evaluate the Performance of High-Throughput Sequencing for Virus Detection” (A Khan AS, Ng SHS, Vandeputte O, Aljanahi A, Deyati A, Cassart JP, Charlebois RL, Taliaferro LP. Wieloośrodkowe badanie oceniające wydajność wysokowydajnego sekwencjonowania do wykrywania wirusów. MSphere. 2017 września 13; 2 (5) .pii: e00307-17. Doi: 10.1128 / mSphere.00307-17. ECollection 2017 wrzesień-październik PubMed PMID : 28932815; PubMed Central PMCID: PMC5597969), podkreślono, w jaki sposób analizy przeprowadzone za pomocą różnych instrumentów HTS, z różnymi protokołami przygotowania próbki i analizy danych, na próbkach naśladujących złożone materiały biologiczne, wykazały podobną czułość w wykrywaniu wirusów w 3 kilka warsztatów.
Wreszcie w „Raporcie z międzynarodowej konferencji na temat sekwencjonowania nowej generacji przypadkowego wykrywania wirusów w produktach biologicznych” (Khan AS, Benetti L, Blumel J, Deforce D, Egan WM, Knezevic I, Krause PR, Mallet L, Mayet D, Minor PD , Neels P, Wang G. Raport z międzynarodowej konferencji na temat sekwencjonowania nowej generacji do przypadkowego wykrywania wirusów w produktach biologicznych. Biologiczne. 2018 września; 55: 1-16. Doi: 10.1016 / j.biologicals.2018.08.002. Epub 2018 6 sierpnia PubMed PMID: 30093175), sprawozdanie z międzynarodowej konferencji, która odbyła się w Rockville (MD) w dniach 26–27 października 2017 r., Współorganizowanej przez IABS (International Alliance for Biological Standardization) i FDA (US Food and Drug Administration), w z udziałem stu dwudziestu ośmiu naukowców z 16 różnych krajów, w tym naukowców Glaxo SmithKline, Merck i Sanofi, stwierdzono, że:
„Głównymi zaletami NGS w testach bezpieczeństwa do wykrywania wirusów jest szybkie i niedocelowe podejście, które można zastosować na wielu podłożach i które pozwala na wykrycie szerokiej gamy wirusów, w tym wariantów i nowych gatunków. ; i że nie ma potrzeby specyficznej amplifikacji, podczas gdy PCR wymaga ukierunkowania na określoną sekwencję ”
I znowu:
„Technologie HTS ujawniają niektóre z obecnych ograniczeń programów do testowania materiałów biologicznych i mogłyby wprowadzić luki w tych programach i zwiększyć bezpieczeństwo produktów”
A we wnioskach raportu czytamy:
„Ciągłe wysiłki w zakresie współpracy i nauki, wymiany między organami regulacyjnymi a innymi organami publicznymi, przemysłem, laboratoriami akademickimi i usługodawcami przyczynią się do rozwoju NGS w celu zagwarantowania bezpieczeństwa produktów ekologicznych wpływających na zdrowie ludzi i zwierząt”
Odpowiadam poniżej na wasze spostrzeżenia, w szczególności na te, które pozwalają nam lepiej wyjaśnić wstępne dane, które opracowaliśmy na temat niektórych partii Priorix Tetra.
Uwaga 1
„Można się zastanawiać, w jaki sposób tę praktykę zachowania tajemnicy, w przeciwieństwie do jakiegokolwiek kryterium udostępniania danych, można pogodzić z ciągłymi żądaniami przejrzystości zgłaszanymi przez stowarzyszenie CORVELVA i prezesa Narodowego Orderu Biologów w zakresie bezpieczeństwa szczepionek: jest to zgodne z prawem uruchomić alarm, nie podając wszystkich danych niezbędnych do oceny jego spójności?
Jak społeczność naukowa i obywatele mogą ocenić wagę niektórych roszczeń, jeśli pełne oryginalne dane są utrzymywane w tajemnicy, do czasu aż prokurator zdecyduje, czy i jak kontynuować postępowanie sądowe? ”
Odpowiedź 1
Dane metagenomiczne ujawnione przez Corvelva dla szczepionki Priorix Tetra (szczepionka MPRV) i omówione w raporcie opublikowanym w grudniu były wstępne (zaktualizowana wersja została ujawniona na stronie internetowej Corvelva 23 stycznia, w wyniku dalszego etapu analizy danych), ponieważ, jak już wielokrotnie powtarzano, ujawnione dotychczas dane wynikają z procesu badań i rozwoju technologicznego rozpoczętego w 2017 r. w celu dopracowania i zoptymalizowania całej procedury, od przetwarzania próbki do analizy bioinformatycznej. Postanowiono ujawnić wyniki w trakcie pracy, gdy będą one dostępne, w absolutnej przejrzystości (wręcz przeciwnie do tego, co określa się jako „praktykę zachowania tajemnicy”!). W każdym razie mocno poparte literaturą naukową na temat argument.
Naszym zdaniem to, co do tej pory ujawniliśmy na temat tej szczepionki, szczególnie w odniesieniu do dużej ilości ludzkiego DNA płodu, nie jest niczym nowym ani nieoczekiwanym, a co najmniej nieznane firmom farmaceutycznym.
Uwaga 2
„Łatwo zauważyć, że suma odczytów wskazanych w lewym górnym rogu (podświetlona na żółto) wynosi ponad 6 milionów; jednakże suma wszystkich odczytów podzielonych według klasy organizmu wynosi około 5 i pół miliona, z „niedoborem” ponad 700.000 88 odczytów (i procentową sumą wskazaną przez czerwoną ramkę, równą XNUMX%). Ten prosty dodatek, powtórzony we wszystkich tabelach, zadaje pytanie, gdzie skończyły się brakujące odczyty (odsetek, który z pewnością nie jest bez znaczenia) i dlaczego nie zostały zgłoszone. Możliwość popełnienia błędu przez badaczy pozostaje otwarta ”
Odpowiedź 2
Powiedzmy, że 700.000 XNUMX odczytów mniej więcej zmienia istotę przeprowadzonych wstępnych analiz przesiewowych. Brakujące sekwencje są uwzględniane przy obliczaniu sekwencji „nieprzypisanych”. Jak już wspomniano, dane nie są ostateczne i mogą występować nieścisłości w raportach. Wszystko będzie sprawdzane i poprawiane w miarę postępu pracy.
Uwaga 3
„Genom wirusa różyczki, jednego z atenuowanych wirusów niezbędnych do nadania immunogenności, nie byłby obecny. Wykrywanie na bardzo niskich poziomach, na granicy szumu statystycznego, mikroorganizmu w badanej próbce nie zależy zatem od jego braku, ale od niezoptymalizowanej metody identyfikacji. Tę samą, identyczną trudność można napotkać przy identyfikacji każdego innego genomu, dla którego metoda analizy nie została odpowiednio skalibrowana; brak ujawnienia genomu wirusa różyczki (ponadto wirusa pojedynczej helisy otoczkowanej) można zatem przypisać zastosowanej metodzie, bez konieczności powoływania się na jej brak w badanych partiach szczepionek. "
Odpowiedź 3
Jego teza jest do przyjęcia. Jesteśmy w fazie badań i rozwoju, jednak technologia ta jest bardzo solidna i uznana przez społeczność naukową zajmującą się głębokim sekwencjonowaniem do wykrywania wirusów w złożonych substratach biologicznych. Testy międzylaboratoryjne i wprowadzenie odpowiednich certyfikowanych standardów mieszanek wirusowych pozwoli nam zweryfikować lub obalić zaproponowaną przez ciebie hipotezę, że metoda nie jest (jeszcze) zoptymalizowana pod kątem wirusów.
W każdym razie genom różyczki został następnie znaleziony w badanej partii, zwiększając głębokość sekwencjonowania (114 sparowanych sekwencji końcowych o długości 125 pz, z około 260 milionów wyprodukowanych sekwencji), jak opisano w drugim raporcie ze stycznia 2019 r. Również w szczepionce MMRVax Pro firmy Merck, analizowany w 2017 r. Tą samą metodą, genom wirusa różyczki został znaleziony bez konieczności wchodzenia na tak głębokie głębokości, co dowodzi, że metoda działa.
Uwaga 4
„Zostałyby wykryte genomy wielu organizmów zanieczyszczających należących do licznych taksonów, w tym robaków pasożytniczych, bakterii i przypadkowych ludzkich wirusów chorobotwórczych. W przedstawionych tabelach pojawia się również kolejny problem, polegający na dużej częstości sekwencji przypisywanych wirusom zintegrowanym z ludzkim genomem (endowirusy o różnej naturze) lub sekwencjom retrowirusowym, o których wiadomo, że można je znaleźć w normalnym ludzkim genomie (na przykład w przypadku sekwencji zintegrowane retrowirusy, które można pomylić z HIV) lub w genomie kurzych komórek embrionalnych wykorzystywanych do produkcji szczepionek, zwykle wykrywane jako fałszywe przypisanie do zanieczyszczających genomów w sekwencjonowaniu DNA lub RNA, w zależności od konkretnej rozważanej endosekwencji. Przede wszystkim istnieje wiele gatunków wymienionych w tabelach raportów, które można znaleźć na podstawie liczby odczytów w pełni uwzględnionych w szumie statystycznym. 3 lub mniej odczytów to ryzykowna procedura, która prowadzi do dużej liczby fałszywie pozytywnych wyników ”
Odpowiedź 4
Wersja raportu ujawniona na stronie internetowej Corvelva w grudniu, do której się odwołujesz, została niedawno zaktualizowana o wersję, w której część zanieczyszczeń została wstępnie zweryfikowana za pomocą alternatywnego oprogramowania, a następnie ręcznie. Pierwsza analiza została celowo przeprowadzona bez filtrów, aby podkreślić każdy możliwy sygnał i wszystkie problemy związane z „otwartą” analizą. Celem naszej pracy nie jest, jak wielokrotnie powtarzano, przeprowadzenie analizy zwolnienia serii, ale przeprowadzenie wstępnego badania przesiewowego, a następnie międzylaboratoryjnego potwierdzenia najważniejszych problemów, które pojawiły się, dzięki zastosowaniu dobrze ugruntowanej technologii w tej dziedzinie. genomowy, już stosowany w szczepionkach, a wkrótce także przez agencje regulacyjne i duże firmy farmaceutyczne w celu podniesienia jakości, a tym samym bezpieczeństwa produktu.
Jeśli zdefiniujesz limit 3 odczytów (w oparciu o co?), To moglibyśmy przynajmniej pomyśleć, że oznaki obecności endogennych retrowirusów w tej szczepionce są prawdopodobne (odczytuje ludzki endogenny retrowirus K: 32 125 pz, co odpowiada 4000 pz i HERVH- env 62: 4 czyta, co odpowiada 500 pz), w danych o sekwencji RNA, które z definicji reprezentują transkrybowany, a zatem potencjalnie aktywny materiał.
Zamiast tego zgadzam się z tobą, że sekwencje retrowirusowe znalezione w danych sekwencji DNA mogą być prowirusami zintegrowanymi z ludzkim genomem, biorąc pod uwagę bardzo dużą ilość ludzkiego DNA w danej szczepionce.
Pamiętam, że w szczepionce Attenuvax (szczepionka przeciw odrze) 4 sekwencje obejmujące 700 pz (Victoria JG, Wang C, Jones MS, Jaing C, McLoughlin K, Gardner S i in. Wirusowe kwasy nukleinowe w żywych atenuowanych szczepionkach: wykrywanie warianty mniejszościowe i przypadkowy wirus J Virol 2010; 84: 6033–40) po raz pierwszy pozwoliły na wykrycie zanieczyszczenia wirusem wirusa białaczki ptasiej (ALV) w szczepionce, a następnie potwierdzono go metodą nested PCR.
Uwaga 5
„Jeśli chodzi o pierwszy z tych potencjalnych zanieczyszczeń, niepatogenną bakterię wiążącą azot Bradyrhizobium, jego„ pojawienie się ”w laboratoriach sekwencjonujących jest dobrze znany jako problem związany z przygotowaniem próbek do sekwencjonowania, przypisywanych zanieczyszczeniu zestawów do oczyszczania DNA, wody i odczynników, które należy zastosować. Dobrze znany problem, w którym w pracy „eksploracyjnej” genomiki próbkę kontrolną, na przykład zawierającą wodę, sekwencjonuje się równolegle z badaną próbką ”
Odpowiedź 5
Białka zostały wykonane (zarówno z ekstrakcji, jak iz biblioteki), ale biblioteki nie zostały uzyskane, dlatego nie można było uruchomić ich w sekwencerze, aby dokładnie sprawdzić, co to jest odczynnik / zanieczyszczenie środowiska. Jednak podejmowane są starania, aby zrozumieć, czym są zanieczyszczenia „fizjologiczne”, a testy międzylaboratoryjne pomogą również w określeniu możliwych zakażeń bakteryjnych zależnych od laboratorium.
Uwaga 6
„Jeśli chodzi o dwa rodzaje robaków ujawnionych w analizie sekwencjonowania RNA, wystarczy zaobserwować, jak nie są one obecne w odpowiednich analizach sekwencjonowania DNA, gdzie powinny być znacznie lepiej wykrywalne; w związku z tym faktyczna obecność tych dwóch gatunków jawi się jako artefakt, którego nie można potwierdzić w świetle przedstawionych danych sekwencjonowania DNA.
W świetle dokonanych rozważań nie jest zatem możliwe potwierdzenie obecności żadnego z zanieczyszczających genomów, a istnieją mocne wskazówki, które skłaniają nas do myślenia o obecności wielu fałszywych wyników pozytywnych ”
Odpowiedź 6
Druga wersja raportu przewidywała już pierwszą walidację niektórych znalezionych organizmów, zarówno z alternatywnym oprogramowaniem, jak i ręcznie.
Ocena nieprawidłowych przypisań oprogramowania Kraken (ponadto jedno z najczęściej używanych programów przez społeczność naukową do analiz metagenomicznych całego genomu) jest wykorzystywana do opracowania doraźnego strumienia bioinformatyki dla tego rodzaju analiz, który nie wymaga użycia innych. oprogramowanie i kontrola manualna (ponadto powszechne w dziedzinie metagenomiki w przypadku problemów ze stanu techniki ze względu na brak konkretnych i szczególnie dokładnych baz danych).
Uwaga 7
„Wykryty ludzki DNA miałby wysoką masę cząsteczkową, a zasięg ludzkiego genomu byłby całkowity, tak że cały genom ludzkich komórek płodowych, a nie jego części, byłby obecny w badanych partiach szczepionek.
„W szczepionce Priorix Tetra ludzkie genomowe DNA ma wysoką masę cząsteczkową (> 10.000 19 pz), a całkowite sekwencyjne pokrycie całego referencyjnego genomu ludzkiego (HG-XNUMX) pokazuje, że jest to cały genom komórek płodu wykorzystywanych do obecność kultury wirusa szczepionkowego, a nie tylko jej części. "
Nie podano bezpośrednich dowodów na to stwierdzenie, bez uszczerbku dla obecności DNA ważącego około 100.000 XNUMX zasad ujawnionego przez elektroforezę żelową przez autorów (udokumentowaną niską jakością w dokumencie CORVELVA, którego wskazane byłoby uzyskanie wysokiej jakości oryginału uchwała w celu oceny, czy dane są prezentowane w nienaruszony sposób).
Jednak autorzy zapominają, że DNA niektórych atenuowanych wirusów obecnych w przedmiotowej szczepionce jest mniej więcej tego samego rozmiaru, co ujawniono w żelu (na przykład DNA wirusa ospy wietrznej wynosi 125 kb); dlatego nawet jeśli chcesz zobaczyć ślady wysokiej wagi DNA na prezentowanym obrazie żelu, można to wyraźnie przypisać DNA atenuowanych wirusów obecnych w szczepionce, a nie fragmentom ludzkiego DNA. Ponownie mamy do czynienia z nadinterpretacją wyniku eksperymentu. Jeśli chodzi o drugi element, na którym opiera się twierdzenie CORVELVA, to znaczy, że cały genom ludzki byłby obecny w szczepionkach, pamiętamy, że nawet nie trzeba mieć dostępu do szczegółowych danych dotyczących zasięgu, aby zważyć jego spójność: zastosowana głębokość sekwencjonowania oraz liczba uzyskanych odczytów to stwierdzenie jest całkowicie nieprawdopodobne. Ponadto zastosowana technika sekwencjonowania, oparta na krótkich sekwencjach, 125 nukleotydach, nie pozwala określić, czy genom jest fragmentowany. Zastosowana technika sekwencjonowania pozwala jedynie oszacować część zsekwencjonowanego genomowego DNA ”
Odpowiedź 7
Ludzkie DNA w tej szczepionce wynosi około 8 do 1 w stosunku do DNA ospy wietrznej (88% odczytów jest pochodzenia ludzkiego, w porównaniu do 11% ospy wietrznej w ostatniej analizowanej partii A71CB256A). Innych atenuowanych wirusów dsDNA nie ma w szczepionce, ponieważ wirusy odry, świnki i różyczki są jednoniciowymi wirusami RNA, których nie można oglądać na żelu agarozowym. NGS jest technologią ilościową, dlatego prosta kwantyfikacja fluorymetryczna całkowitego DNA wyekstrahowanego ze szczepionki (np. Partia. A71CB256A = 3,7 mikrograma na dawkę), związana z rozważeniem względnej kwantyfikacji wykonanej powyżej (8: 1), pozwala nam móc powiedzieć, że komórkowy DNA wynosi około 2,9 mikrograma na dawkę, w porównaniu do około 0.74 mikrograma DNA ospy wietrznej. Jest zatem prawdopodobne, że co najmniej część DNA o wysokiej masie cząsteczkowej widoczna na żelu może być komórką płodowej linii komórkowej MRC5.
Bezpośredni dowód na to, że w tym produkcie znajduje się KOMPLETNY genom ludzki (tj. Z niekodującymi genami i sekwencjami), pofragmentowany lub nie, jest podany w wyniku dopasowania odczytów pochodzących od człowieka w odniesieniu do ludzkiego odniesienia hg19.
Poniższa tabela, zaznaczona na pomarańczowo, przedstawia wynik wyrażony jako „Av_cov = średnie pokrycie” przyrównania ludzkich sekwencji z 3 badanych partii Priorix (pierwsza, druga i trzecia partia) na ludzkich chromosomach. W kolumnie 1 chrM to mitochondrialne DNA, podczas gdy Chr2 do ChrY to złożone ludzkie chromosomy, w tym chromosomy płciowe X i Y. Kolumna 3 przedstawia długość złożonych ludzkich chromosomów wyrażonych w parach zasad.
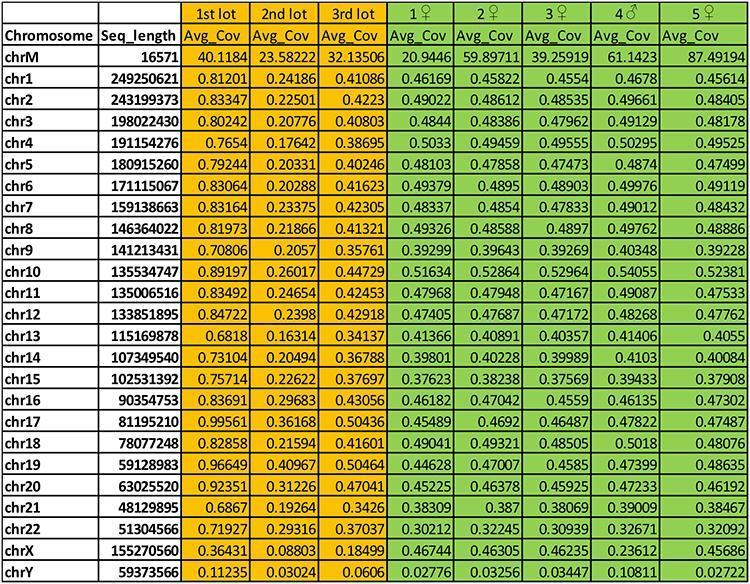
Pokrycie jest niskie (średnio Avg_cov wzdłuż każdego chromosomu <niż 1x), ale jednorodność rozmieszczenia odczytów, które są jednoznacznie wyrównane, wzdłuż wszystkich ludzkich chromosomów, a także obecność odczytów, które są zgodne z wyższym pokryciem na genomie mitochondrialne (w ludzkich komórkach są 2 genomy, jeden jądrowy o wielkości około 3 pb i podzielony na chromosomy i jeden mniejszy, okrągły, mitochondrialny o wielkości około 16 kpz), pozwala nam jednak bezsprzecznie rozpoznać sytuację podobną do dolnoprzepustowego sekwencjonowania genomu ludzkiego genomu indywidualny. Dla łatwiejszego zrozumienia tego, co zostało powiedziane, w części zaznaczonej na zielono istnieje kilka dolnoprzepustowych sekwencjonowania całego genomu ludzkiego (5 próbek, 4 kobiety i 1 mężczyzna) na głębokości podobnej do tej uzyskanej dla 3 partii szczepionek Priorix Tetra.
Wskazanie płci osoby (najprawdopodobniej mężczyzny) w szczepionce można również wywnioskować ze związku między średnim pokryciem chromosomu X i Y.
Ludzki DNA zawarty w uprzednio zsekwencjonowanych partiach Priorix został również zakwalifikowany jako należący do linii płodowej MRC-5, czyli ciągłej linii komórkowej z tkanki płucnej męskiego płodu poronionego w latach 60., w którym wirus ospa wietrzna i różyczka. Analiza wariantów mitochondrialnego DNA obecnego w szczepionce w porównaniu z mitochondrialnym DNA linii MRC-5 (DNA linii komórkowej zakupiono od ATCC, głównego zasobu standardowych materiałów biologicznych na świecie) pokazuje, że są one tą samą osobą.
Uwaga 8
„Od dawna istnieją dowody, że nawet bardzo duże dawki ludzkiego DNA nie mogą powodować znaczącego ryzyka nawet w długich okresach obserwacji, co potwierdzają badania na naczelnych innych niż ludzie, na podstawie doświadczeń klinicznych ze szczepionkami i innymi lekami biologicznymi oraz z ostatnie badania oceny ryzyka co sugeruje, że nawet w krajach pozaeuropejskich, w których istnieją progi ilości DNA do wstrzykiwań, progi te są niepotrzebnie rygorystyczne ”
Odpowiedź 8
Artykuł z 1995 r. (https://www.sciencedirect.com/science/article/pii/S104510568570036X?via%3Dihub), o którym wspomina Pan, dotyczy badania naczelnych, które moim zdaniem z pewnością nie wystarcza do wykazania braku zakaźności, onkogenności i autoimmunizacji ludzkiego DNA wstrzykniętego dzieciom, jak stwierdzili ci sami autorzy w końcowej części dyskusji. W eksperymencie dużą ilość ludzkiego DNA wstrzykuje się małpom, a zatem nie DNA tego samego gatunku.
Drugi cytowany artykuł (https://www.ncbi.nlm.nih.gov/pubmed/23569076) stwierdza bardzo wyraźnie, że resztkowe DNA jest zakaźne przy 2 mikrogramach, nawet jeśli w każdym razie limit ten jest uzyskiwany z oceny statystycznej, a nie z danych eksperymentalnych. W każdym razie, zgodnie ze wskazaniami autora, szczepionka Priorix miałaby pewną ilość DNA płodu wyraźnie zgodną z tą określoną jako zagrożoną chorobą zakaźną.
Merck dla szczepionki Varivax (ospa wietrzna, żywy atenuowany wirus) deklaruje w amerykańskiej ulotce obecność ludzkiego płodowego DNA pochodzącego z komórek MRC-5, który z analiz przeprowadzonych przez dr Deishera wydaje się być w ilości około 2 mikrogramów.
W przypadku włoskiej ulotki Priorix Tetra obecność DNA linii MRC-5 nie jest jednak wskazana, mimo że jest w ilościach tego samego rzędu wielkości, co w amerykańskim Varivax. W każdym razie 2 mikrogramy płodowego DNA MRC-5 nie są moim zdaniem resztkową ilością, ale prawdziwym składnikiem szczepionki.
Badania Dr. Deishera in vitro, które, mamy nadzieję, zostaną wkrótce opublikowane, niestety pokazują zaskakujące wyniki, gdy szczepionkę zawierającą ludzki DNA płodu (hipometylowany) inkubuje się in vitro z ludzkimi hematopoetycznymi komórkami macierzystymi. Ale czekamy na opublikowanie badań, a następnie możemy ponownie spotkać się na ten temat.
Loretta Bolgan *
* Doktor chemii i technologii farmaceutycznych, doktorat z nauk farmaceutycznych na uniwersytecie medycznym Harvard w Bostonie. Pracowała w sektorze przemysłu farmaceutycznego, gdzie zajmowała się rejestracją i rozwojem projektów badawczych w dziedzinie onkologii. Konsultant części ustawy 210/92 dotyczącej zanieczyszczenia środowiska i chorób zawodowych, uczestniczył w ostatniej sejmowej komisji śledczej w sprawie zubożonego uranu w grupie szczepionek. Obecny konsultant Narodowego Zakonu Biologów ds. Toksykologii leków i szczepionek, zajmuje się również odżywianiem i terapiami uzupełniającymi.